Beginning with the discovery of penicillin by Alexander Fleming in the late 1920s, antibiotics have revolutionized the field of medicine. They have saved millions of lives each year, and have even been used prophylactically for the prevention of infectious diseases. However, we have now reached a crisis where many antibiotics are no longer effective. Such infections often result in an increased number of hospitalizations, more treatment failures and the persistence of drug-resistant pathogens.

Better understanding of mechanisms of antibiotic resistance would allow the development of control strategies to reduce the spread of resistant bacteria and its evolution. Bacteria may be intrinsically resistant to a class of antibiotics or may acquire resistance.
Main mechanisms of resistance to antibiotics can be caused by:
Alteration of the target site of the antibiotic. A common strategy for bacteria to develop antimicrobial resistance is to avoid the action of the antibiotic by interfering with their target site. To achieve this, bacteria have evolved different tactics, including protection of the target (avoiding the antibiotic to reach its binding site) and modifications of the target site that result in decreased affinity for the antibiotic molecule.
Enzyme inactivation of the antibiotic. One of the most successful bacterial strategies to cope with the presence of antibiotics is to produce enzymes that inactivate the drug by adding specific chemical moieties to the compound or that destroy the molecule itself, rendering the antibiotic unable to interact with its target.
Active transport of the antibiotic out of the bacterial cell. The production of complex bacterial machineries capable to extrude a toxic compound out of the cell can also result in antimicrobial resistance. Many classes of efflux pumps have been characterized in both Gram-negative and Gram-positive pathogens.
Decreased permeability of the bacterial cell wall to the antibiotic. Many of the antibiotics used in clinical practice have intracellular bacterial targets or, in case of Gram-negative bacteria, located in the cytoplasmic membrane (the inner membrane). Therefore, the compound must penetrate the outer and/or cytoplasmic membrane in order to exert its antimicrobial effect. Bacteria have developed mechanisms to prevent the antibiotic from reaching its intracellular or periplasmic target by decreasing the uptake of the antimicrobial molecule.

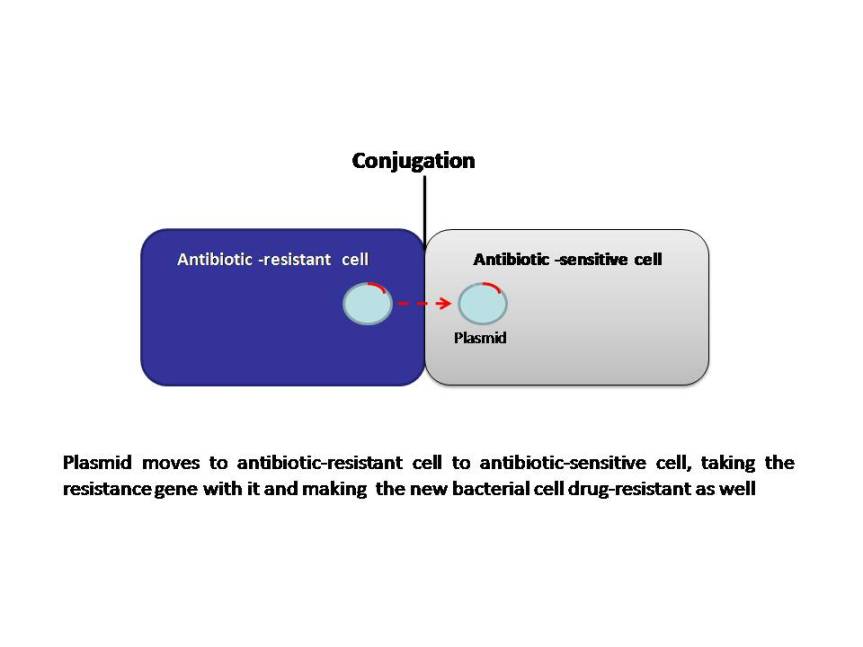
Bacteria can develop resistance to antibiotics by mutating existing genes (vertical evolution), or by acquiring new genes from other strains or species (horizontal gene transfer). Many of the antibiotic mobile resistance genes (MGEs) are carried on genetic elements (plasmids, transposons or phages) that act as vectors that transfer these genes to other members of the same bacterial species, as well as to bacteria in another genus or species. The MGEs allow resistance to spread horizontally and disseminate among different bacterial species. Although this association seems improbable, it appears to occur frequently and follows a series of evolutionary steps fueled by natural selection (antibiotic selection). The power of modern DNA sequence analysis allows us to better understand the process of emergence of these genetic structures. The primary mechanisms of horizontal transfer are conjugation (plasmids are transferred from a donor cell to a recipient cell), transformation (uptake of naked DNA), and transduction (bacteriophages as transporters of genetic information). Conjugation is considered the principal mode for antibiotic resistance transfer, because many resistance genes are situated on mobile elements, such as plasmids integrons and transposons.
Most families of antibiotics present in nature are compounds produced by fungi or bacteria; bacteria utilize these compounds to eliminate competitor microorganisms. As part of this arms race, many microorganisms code for genes whose products neutralize antibiotics; these genes may have been present in bacterial chromosomes for millions of years and they were probably not mobile, as evidenced by recent findings. The massive use of antibiotics probably favored selection of antibiotic resistant bacteria resulting in large numbers of bacteria coding for resistance genes. On the other hand, bacterial chromosomes are populated with transposable elements (insertion sequences known as ISs), which jump frequently and randomly, as demonstrated during in vitro experiments from one genetic location to another in the same cell. Antibiotic resistance genes could be mobilized to genetic structures, such as plasmids and phages, which can move horizontally between bacterial cells including different bacterial species. The association of resistance genes to these mobile structures could occur through ISs; this has been postulated as the origin of many MGEs. Alternatively, plasmids or phages may also integrate in the bacterial chromosome in the vicinity of resistance genes and then mobilize the resistance genes as these structures excise from chromosomes.
Emergence of resistance among the most important bacterial pathogens is recognized as a major public health threat affecting humans worldwide. Widespread use of antibiotics has undoubtedly facilitated the development of antimicrobial resistance worldwide. Better understanding of the mechanisms of antibiotic resistance, will help clinicians regarding usage of antibiotics in different situations.
Antibiotic resistance in Enterobacteriaceae
Resistance to beta-lactam agents
Beta-lactam antibiotics exhibit the most common treatment for bacterial infections and continue to be the prominent cause of resistance to beta-lactam antibiotics among Gram-negative bacteria worldwide. The persistent exposure of bacterial strains to a multitude of beta-lactams has induced dynamic and continuous production and mutation of beta-lactamases in these bacteria, expanding their activity against most beta-lactam antibiotics. Resistance to beta-lactams in Enterobacteriaceae is mainly conferred by beta-lactamases. These enzymes inactivate beta-lactam antibiotics by hydrolysis. Beta-lactamases are commonly classified according to two systems: the Ambler molecular classification and the Bush–Jacoby–Medeiros functional classification. The Ambler scheme classifies beta-lactamases into four classes according to the protein homology of enzymes. Beta-lactamases of class A, C, and D are serine beta-lactamase and class B enzymes are metallo-beta-lactamases. The Bush–Jacoby–Medeiros functional scheme is based on functional properties of enzymes and on their ability to hydrolyze specific beta-lactam classes. This classification was updated in 2010. The updated system includes group 1 (class C) cephalosporinases; group 2 (classes A and D) broad-spectrum, inhibitor-resistant, extended-spectrum beta-lactamases and serine carbapenemases; and group 3 (class B) metallo-beta-lactamases.
Group 1 enzymes are cephalosporinases belonging to molecular class C. They are more active on cephalosporins than benzylpenicillin. It includes AmpC beta-lactamases. AmpC-hyperproducing mutants are resistant to penicillins, aztreonam, third generation cephalosporins including cefotaxime, ceftazidime, ceftriaxone and even ertapenem when the enzyme is massively expressed. Cefepime, a fourth-generation cephalosporin with broader spectrum activity compared to ceftriaxone, is a poor inducer of AmpC beta-lactamase. Many AmpC-producing organisms are susceptible to cefepime because cefepime is poorly hydrolyzed by the AmpC beta-lactamase enzyme. However, the role of cefepime in treating infections caused by AmpC-producing organisms is controversial because of the inoculum effect.
Group 2 (classes A and D) represent the largest group of beta-lactamases, it includes ESBL producing Enterobacteriaceae and carbapenemases (class A) and OXA beta-lactamases (class D). ESBL are enzymes capable of hydrolyzing and inactivating a wide variety of beta-lactams, including third-generation cephalosporins, penicillins, and aztreonam. Most ESBLs of clinical interest are encoded by genes located on plasmids. These plasmids may also carry genes encoding resistance to other multiple drug classes including aminoglycosides and fluoroquinolones. The main ESBL enzymes imparting antibiotic resistance are TEM-, SHV-, and CTX-M. Although hyperproduction of beta-lactamases or additional resistance mechanisms may hamper the antibiotic effectiveness, most TEM, SHV and CTX-M variants remain susceptible in vitro to the “old” beta-lactam/beta-lactam inhibitor combinations such as piperacillin-tazobactam. However, the efficacy of piperacillin-tazobactam for treating serious ESBL infections is controversial. Yet, there have been concerns that in vitro susceptibility may not reliably translate into clinical efficacy. This has been based largely on concerns over inoculum effects, the co-location of other beta-lactamase enzymes (which may not be well inhibited by beta-lactamase inhibitors) on acquired plasmids and the potential for additional resistance mechanisms such as alterations in outer membrane proteins. Furthermore, in critically ill patients the pharmacokinetic properties of beta-lactam agents are modified and these patients may have adverse outcomes as a result of sub-optimal antibiotic exposure. Rates of CTX-M infections have increased during the last decade compared with rates of TEM- and SHV- infections. The diffusion of CTX-M-producing Enterobacteriaceae are common in Southeast Asia and Eastern Mediterranean countries. The OXA-type beta-lactamases are so named because of their oxacillin-hydrolyzing abilities. OXA beta-lactamases have resistance limited to the penicillins, but some became able to confer resistance to cephalosporins. OXA-1 and OXA-10 beta-lactamases have only a narrow hydrolytic spectrum. However, other OXA beta-lactamases including OXA-11, -14, -15, -16, -28, -31, -35 and -45 confer resistance to cefotaxime, ceftazidime and aztreonam. OXA-23 and OXA-48 are classes of carbapenemases that belong to OXA-type beta-lactamases with carbapenem-hydrolyzing activities. While OXA-23 appears most frequently in A. baumannii, OXA-48 enzymes have now become widespread in the Enterobacteriaceae, especially in Mediterreanean countries. Klebsiella pneumoniae carbapenemases (KPCs) are beta-lactamases produced by Gram-negative bacteria. They efficiently hydrolyse penicillins, all cephalosporins, monobactams, beta-lactamase inhibitors, and even carbapenems. KPCs are becoming an increasingly significant problem worldwide. KPC-producing K. pneumoniae pose a serious threat in clinical situations where administration of effective empiric antibiotics is essential to prevent mortality following bacteraemia and infections in immunocompromised patients including organ transplant recipients and those with cancer.
Group 3 (Class B) metallo-beta-lactamases (MBLs) differ structurally from the other beta-lactamases by their requirement for a zinc ion at the active site. They are all capable of hydrolysing carbapenems. In contrast to the serine beta-lactamases, the MBLs have poor affinity or hydrolytic capability for monobactams and are not inhibited by clavulanic acid or tazobactam. The most common metallo-beta-lactamase families include the IMP, VIM and NDM.
Two new cephalosporin beta-lactamase inhibitor combinations, ceftolozane/tazobactam and ceftazidime/avibactam, have been approved recently for the treatment of patients with complicated intra-abdominal infections. Ceftolozane/tazobactam is active in vitro against many ESBL-producing Enterobacteriaceae, It is also active against many strains of P. aeruginosa and appears to be the most potent currently available beta-lactam or beta-lactam-beta-lactamase inhibitor combination against this organism. Ceftazidime/avibactam has activity against most strains of Enterobacteriaceae, including ESBL-producing strains and AmpC beta-lactamase-producing strains. Ceftazidime/avibactam is the only currently available beta-lactam-beta-lactamase inhibitor combination with substantial in vitro activity against KPC-producing Enterobacteriaceae. Both ceftolozane/tazobactam and ceftazidime/avibactam are not active against MBL-producing micro-organisms.
Antibiotic resistance to fluoroquinolones
All Enterobacteriaceae are naturally susceptible to fluoroquinolones. The process by which susceptible strains become highly fluoroquinolone resistant is thought to be a result of a series of sequential steps and several mutations are needed to produce a high level of fluoroquinolone resistance. High-level resistance emerges after successive chromosomal mutations in the DNA gyrase- encoding gyrA gene and topoisomerase IV-encoding parC gene. The over-expression of efflux pumps may also play a role in the high level of resistance in certain strains. Resistance to fluoroquinolones can also be mediated by the plasmid-encoded qnr genes, which confer protection of bacterial topoisomerases against fluoroquinolones, the plasmid-encoded efflux pump qepA.
Resistance to aminoglycosides
Aminoglycosides resistance occurs through several mechanisms that can simultaneously coexist. Aminoglycosides resistance in Enterobacteriaceae relies mainly on the genes encoding aminoglycoside-modifying enzymes (AMEs). AMEs hamper antibiotic activity. AMEs are often located on plasmids that carry multiple resistance genes, including ESBL Other described mechanisms of resistance include modification of the antibiotic target by mutation of the 16S rRNA or ribosomal proteins, methylation of 16S rRNA (by RNA methlysases which genes are often co-located with beta-lactamase encoding genes), reduced permeability and/or increasing active efflux of the antibiotic.
Antibiotic resistance in non-fermenting gram-negative bacteria
Non-fermenting Gram-negative bacteria (P. aeruginosa, S. maltophilia and A. baumannii) are intrinsically resistant to many drugs and can acquire resistance to virtually any antimicrobial agent. A variety of resistance mechanisms have been identified in P. aeruginosa and other Gram-negative non-fermenting bacteria, including impermeable outer membranes, expression of efflux pumps, target alteration and production of antibiotic-hydrolyzing enzymes such as AmpC beta-lactamases that are either chromosomally encoded or acquired. These mechanisms may be present simultaneously, conferring multiresistance to different classes of antibiotics. These mechanisms may also allow transmission to multiple strains of bacteria. P. aeruginosa is intrinsically resistant to a number of beta-lactam antibiotics including amoxicillin, first and second generation cephalosporins, cefotaxime, ceftriaxone and ertapenem. Effective agents include ticarcillin, piperacillin, ceftazidime, cefepime, imipenem, meropenem and doripenem. Aztreonam activity is variable. Unlike tazobactam, clavulanate is a strong inducer of AmpC in P. aeruginosa. P. aeruginosa also has the ability to acquire beta-lactamases, including ESBL and carbapenemases. The P. aeruginosa genome contains several different multidrug resistance efflux pumps, which reside in the membrane and remove antimicrobials and toxins, thereby lowering their concentration inside the cell to sub-toxic levels. Overproduction of these pumps reduces susceptibility to a variety of antibiotics. The most common system is MexAB-OprM. Its overexpression confers resistance to ticarcillin, aztreonam, and at a lesser extent, meropenem. Reduced outer-membrane permeability caused by qualitative or quantitative alterations of the OprD porin, which manages the passage of imipenem through the outer membrane, confers P. aeruginosa a basal level of resistance to carbapenems, especially to imipenem. The mechanisms of AMR in A. baumannii are various, and generally include production of beta-lactamases, impermeable outer membrane, expression of efflux pumps, and change of targets or cellular functions such as alterations in penicillin-binding proteins (PBPs). The PBPs play a crucial role in the synthesis of peptidoglycan, an essential component of the bacterial cell wall. A. baumannii naturally produces a non-inducible AmpC-type cephalosporinase (ACE-1 or ACE-2) and an OXA-51-like carbapenemase which confers, at basal levels of expression, intrinsic resistance to aminopenicillins, first and second generation cephalosporins and aztreonam. Ertapenem naturally lacks activity against non-fermenting Gram negative bacteria including A. baumannii. Overproduction of the AmpC-type cephalosporinase confers acquired resistance to carboxypenicillins, ureidopenicillins and third generation cephalosporins. The emergence of carbapenem-resistant clones of A baumannii has been reported since the late 1980s. Carbapenem resistance can result from the over-expression of OXA-51-like oxacillinase, and from the acquisition of OXA-23-like, IMP, VIM, SIM or, more recently, NDM-type carbapenemases. Acquired resistances to fluoroquinolones (mutations in gyrA and/or parC) and aminoglycosides (plasmid-borne AMEs) may be observed in ESBL as well as carbapenemase-producing A. baumannii strains. Colistin resistant isolates are now increasing worldwide. Resistance to colistin is thought to be mediated by modifications of the lipopolysaccharides of the bacterial cell membrane that interfere with the agent’s ability to bind bacterial targets.
Antibiotic resistance in enterococci
Enterococci are intrinsically resistant to some penicillins, all cephalosporins, and, at a low level, to aminoglycosides. Additionally, they have acquired resistance to many other classes of antibiotics. Enterococci have intrinsic resistance to most beta-lactam antibiotics because of the low affinity penicillin binding proteins (PBPs). Attachment of beta-lactam agents to PBPs results in impaired cell wall synthesis and, in most cases, programmed cell death via creation of reactive oxygen species. Enterococci express low-affinity PBPs (PBP5 in E. faecium, PBP4 in E. faecalis) that bind weakly to beta-lactam antibiotics. Enterococci may develop increased resistance to penicillins through acquisition of beta-lactamases (very rare) or PBP4/5 mutations. Higher level of resistance in E. faecium has been attributed to over production of low affinity PBP-5, a protein that can take over the function of all PBPs In addition, enterococci are “tolerant” to the activity of beta-lactams, and may appear susceptible in vitro but develop tolerance after exposure to penicillin. This property is an acquired characteristic. Enterococci quickly develop tolerance after exposure to as few as five doses of penicillin. Enterococci exhibit intrinsic low-level resistance to all aminoglycosides, precluding their use as single agents. Intrinsic resistance is attributed to an inability of the aminoglycoside to enter the cell (where they act by inhibiting ribosomal protein synthesis). While intrinsic mechanisms result in low-level aminoglycoside resistance, acquisition of mobile genetic elements typically underlies high-level aminoglycoside resistance in both E. faecium and E. faecalis. High-level resistance most frequently occurs through acquisition of a bifunctional gene encoding aph(2′′)-Ia-aac(6′)-Ie, which inactivates aminoglycosides. However, several other genes have been identified that confer gentamicin resistance, including aph(2′′)-Ic, aph(2′′)-Id and aph(2′′)-Ib. These genes are minor contributors to resistance compared to aph(2′′)-Ia-aac(6′)-Ie. Their prevalence varies by geographical region. The acquisition of glycopeptides resistance by enterococci has seriously affected the treatment and control of these organisms. Glycopeptides act by binding to the pentapeptide precursors of enterococci, thereby inhibiting cell wall synthesis. Glycopeptide-resistant organisms modify these pentapeptide precursors, which bind glycopeptides with 1000-fold lower affinity than normal precursors. Various phenotypes of vancomycin-resistant enterococci (VRE) have been characterized; VanA and VanB operons are by far the most prevalent in human glycopeptide-resistant enterococci (GRE) infections. GRE have emerged as a major cause of nosocomial infections. The majority of GRE infections have been attributed to E. faecium, though glycopeptide resistance occurs in E. faecalis and other Enterococcus species as well.
Antibiotic resistance in Staphylococcus aureus
Staphylococcus aureus is a major human pathogen worldwide. Methicillin-resistant S. aureus (MRSA) poses a significant and enduring problem to the treatment of infection by such strains. Resistance is usually conferred by the acquisition of a nonnative gene encoding a penicillin binding protein (PBP2a), with significantly lower affinity for β-lactams. This resistance allows cell-wall biosynthesis, the target of beta-lactams, to continue even in the presence of typically inhibitory concentrations of antibiotic. PBP2a is encoded by the mecA gene, which is carried on a distinct mobile genetic element (SCCmec), the expression of which is controlled through a proteolytic signal transduction pathway comprising a sensor protein (MecR1) and a repressor (MecI).
S. aureus strains exhibiting increased resistance to vancomycin, known as vancomycin intermediate-resistant S. aureus (VISA) were discovered in the 1990s. The molecular basis of resistance in VISA is polygenic and involves stepwise mutations in genes encoding molecules predominantly involved in cell envelope biosynthesis. S. aureusi solates with complete resistance to vancomycin (MIC ≥ 16 µg/mL) are termed vancomycin-resistant S. aureus (VRSA)-they were first reported at the beginning of 2000s. Complete vancomycin resistance in S. aureus is conferred by the vanA operon encoded on transposon Tn1546, originally a part of a vancomycin-resistant enterococci (VRE) conjugative plasmid. S. aureus can acquire enterococcal plasmids during discrete conjugation events. Vancomycin resistance in S. aureus is maintained by retaining an original enterococcal plasmid or by a transposition of Tn1546 from the VRE plasmid into a staphylococcal resident plasmid
Reference
Sartelli M, Weber DG, Ruppé E, Bassetti M, Wright BJ, Ansaloni L, et al. Antimicrobials: a global alliance for optimizing their rational use in intra-abdominal infections (AGORA). World J Emerg Surg. 2016 Jul 15;11:33.
